Introduction
The input format
The pattern matching algorithm
The scan option
The advanced scan option
The SH3-Hunter output
The SH3-Hunter engine
References
Introduction
SH3-Hunter is a web server aimed at identifying SH3 domain interaction sites on protein sequences. It can be used straightforwardly by submitting one ore more protein sequences or a list of peptides and obtaining in output a list of significant interaction sites for the binding of a pre-compiled list of SH3 domains (see Table H1). This is the simple scan mode, in which all the sequences submitted in input are analyzed with respect to all available SH3 domains. An advanced scan mode is also possible, where users can submit the sequence of an SH3 domain and/or select one or more SH3 domains and verify the interactions between them and the submitted sequences. The user SH3 domain can be inserted through an appropriate input text line just above the pre-compiled list of domains. The selection of SH3 domains and query sequences can be made via appropriate checkboxes.
The output page consists of a list of possible interactors sites identified in the submitted sequences and that SH3-Hunter considers as significant. The full set of results (even the domain-peptide pair predictions that the server considers as non significant) can be downloaded as a text file. Each site is proposed as possible binder against the complete SH3 domain list (scan mode) or by a selection of SH3 domains (advanced scan mode) and the corresponding peptide-domain pairs are evaluated by a significance prediction score and a level of sensitivity and precision. These three indicators are the measures of the prediction reliability. The predicting tool is a neural network that integrates both sequence and structure information of the peptide-domain pair, involving a knowledge-based numerical encoding of the input information (Ferraro et al. 2006). The neural network is trained by a dataset of experimentally verified interacting and non-interacting peptide-domain pairs (Landgraf et al. 2004; Tong et al. 2002). In the following, each section of the server is explained in detail. Also the background methodology of the neural network predictor is briefly summarized.
top
The input format
The accepted input types are (all types are case insensitive):
1) FASTA
>sp|P12931|SRC_HUMAN PROTO-ONCOGENE TYROSINE-PROTEIN KINASE SRC (EC 2.7.1.112) (P60-SRC) (C-SRC) - Homo sapiens (Human). GSNKSKPKDASQRRRSLEPAENVHGAGGGAFPASQTPSKPASADGHRGPS AAFAPAAAEPKLFGGFNSSDTVTSPQRAGPLAGGVTTFVALYDYESRTET DLSFKKGERLQIVNNTEGDWWLAHSLSTGQTGYIPSNYVAPSDSIQAEEW YFGKITRRESERLLLNAENPRGTFLVRESETTKGAYCLSVSDFDNAKGLN VKHYKIRKLDSGGFYITSRTQFNSLQQLVAYYSKHADGLCHRLTTVCPTS KPQTQGLAKDAWEIPRESLRLEVKLGQGCFGEVWMGTWNGTTRVAIKTLK PGTMSPEAFLQEAQVMKKLRHEKLVQLYAVVSEEPIYIVTEYMSKGSLLD FLKGETGKYLRLPQLVDMAAQIASGMAYVERMNYVHRDLRAANILVGENL VCKVADFGLARLIEDNEYTARQGAKFPIKWTAPEAALYGRFTIKSDVWSF GILLTELTTKGRVPYPGMVNREVLDQVERGYRMPCPPECPESLHDLMCQC WRKEPEERPTFEYLQAFLEDYFTSTEPQYQPGENL
The first row must contain the initial character '>' followed by a text identifying the sequence.
Different sequences have to be identified by different names.
It is recommended that all lines of text be shorter than 80 characters in length.
Blank lines are not allowed in the middle of FASTA input.
2) Bare Sequence
This may be just lines of sequence data e.g.:
GSNKSKPKDASQRRRSLEPAENVHGAGGGAFPASQTPSKPASADGHRGPSAAFAPAAAEPKLFGGFNSSDTVTSPQRAGPLAGGVTTFVALYDYESRTET
DLSFKKGERLQIVNNTEGDWWLAHSLSTGQTGYIPSNYVAPSDSIQAEEWYFGKITRRESERLLLNAENPRGTFLVRES
ITSRTQFNSLQQLVAYYSKHADGLCHRLTTVCPTSKPQTQGLAKDAWEIPRESLRLEVKLGQGCFGEVWMGTWNGTTRVAIKTLK
PGTMSPEAFLQEAQVMKKLRHEKLVQLYAVVSEEPIYIVTEYMSKGSLLD
Important: Multiple sequences must be separated by a blank line.
3) GenBank/GenPept flatfile format
1 qikdllvsss tdldttlvlv naiyfkgmwk tafnaedtre mpfhvtkqes kpvqmmcmnn 61 sfnvatlpae kmkilelpfa sgdlsmlvll pdevsdleri ektinfeklt ewtnpntmek 121 rrvkvylpqm kieekynlts vlmalgmtdl fipsanltgi ssaeslkisq avhgafmels 181 edgiemagst gviedikhsp eseqfradhp flflikhnpt ntivyfgryw spImportant: Multiple sequences must be separated by a blank line.
4) SwissProt flatfile format
1 MMKRQLHRMR QLAQTGSLGR TPETAEFLGE DLLQVEQRLE PAKRAAHNIH KRLQACLQGQ 60
61 SGADMDKRVK KLPLMALSTT MAESFKELDP DSSMGKALEM SCAIQNQLAR ILAEFEMTLE 120
121 RDVLQPLSRL SEEELPAILK HKKSLQKLVS DWNTLKSRLS QATKNSGSSQ GLGGSPGSHS 180
181 HTTMANKVET LKEEEEELKR KVEQCRDEYL ADLYHFVTKE DSYANYFIRL LEIQADYHRR 240
241 SLSSLDTALA ELRENHGQAD HSPSMTATHF PRVYGVSLAT HLQELGREIA LPIEACVMML 300
301 LSEGMKEEGL FRLAAGASVL KRLKQTMASD PHSLEEFCSD PHAVAGALKS YLRELPEPLM 360
361 TFDLYDDWMR AASLKEPGAR LQALQEVCSR LPPENLSNLR YLMKFLARLA EEQEVNKMTP 420
421 SNIAIVLGPN LLWPPEKEGD QAQLDAASVS SIQVVGVVEA LIQSADTLFP GDINFNVSGL 480
481 FSAVTLQDTV SDRLASEELP STAVPTPATT PAPAPAPAPA PAPALASAAT KERTESEVPP 540
541 RPASPKVTRS PPETAAPVED MARRTKRPAP ARPTMPPPQV SGSRSSPPAP PLPPGSGSPG 600
601 TPQALPRRLV GSSLRAPTVP PPLPPTPPQP ARRQSRRSPA SPSPASPGPA SPSPVSLSNP 660
661 AQVDLGAATA EGGAPEAISG VPTPPAIPPQ PRPRSLASET N
Important: Multiple sequences must be separated by a blank line. The possible 'X' character
in the protein sequences are ignored.top
The pattern matching algorithm
The submission of the sequence implies a first scanning by a pattern matching algorithm in order to verify the presence of proline-rich sites. The presence of proline-rich sites is considered by the server as a mandatory condition for a protein to be a possible interactor of an SH3 domain. The server provides two types of pattern matching filter: The default one search peptides conforming to the standard class I ([RKHFWY]xxPxxP) or class II (PxxPx[RKH]) SH3 binding motifs; a more relaxed filter is proposed in order to extend the peptide space search to those peptides showing only the PxxP proline-rich core. For each match of this last filter, two peptides of 10 residues are identified, one for the class I binding orientation (the one with PxxP at C-term) and one for the class II binding orientation (the one with PxxP at N-term). The two peptides are analyzed by the server separately. SH3-Hunter analyzes only putative interaction sites that agree with the poly-proline motifs. The application of such a filtering procedure maintains a high level of reliability of the server predictions and is based on the fact that the neural predictor is trained by class I and class II interacting data. Specifically, the default filter makes the server prediction more reliable than the relaxed filter. Any sequence that does not contain one of these consensi will be considered as non-interacting.
The pattern matching algorithm permits the extraction from the submitted sequence of short peptides (10 amino acids long) that are then scanned by the neural predictor. Each submitted sequence is, therefore, analyzed as a list of putative interacting peptides.
top
The scan option
In the scan option, SH3-Hunter first applies the pattern matching algorithm and then it establishes all the possible pairing between the matching peptides and the complete SH3 domains server list (Table H1). The resulting peptide-domain pairs represent an exhaustive exploration of the full interaction network between the submitted proteins and the available SH3 domains. The corresponding output page is quickly available and only shows the significant interaction pairs.
top
The advanced scan option
The advanced scan allows users to focus on specific SH3 domains in order to explore the interaction specificity of single domains. Such mode involves an intermediate input page where users can either submit the sequence of their own SH3 domain and/or select one or more SH3 domains from the available list. Domains can be selected by checking the corresponding boxes on the right side of the list. Each user's submitted sequence is represented as a list of peptides conforming SH3 binding motifs. Users can also select a part of the available groups of peptides, thus refining their own submission. Each group of peptides can be selected by clicking on the corresponding checkboxes.
The submission of a new SH3 domain by the user requires the identification of surface contact positions on the domain sequence (see below, The SH3-Hunter engine and Ferraro et al., 2006). This can be achieved in a first approximation by aligning the user domain sequence to the profile generated from the multiple sequence alignment of the SH3 domains in table Table H1. Such profile was carefully generated taking into account the structural alignment of SH3 domains with known 3D structure. All the domains belonging to Table H1 contribute to the definition of the profile. However, the inference of contact positions of the user domain is based only on domain sequence information and thus, at this stage, it must be considered as characterized by a lower reliability.
After the selection of SH3 domains and groups of peptides, a scan button just below the list of peptides permits the final submission. The peptide-domain pairs thus formed represents all the possible combinations between the selected peptides and the selected domains. The corresponding output page provides the list of the significant interacting pairs.
top
The SH3-Hunter output
The results of the proteins scanning appear in the output page, which is organized as a table and reports several information in different columns: The input sequences, with the identifiers on top and the matching peptides highlighted in colour (blue for class I, red for class II) along the sequence; the coloured peptides extracted from each submitted sequence, with the range that defines their position in the sequence; the clickable domain name, that identifies the interaction partner of the corresponding peptide (the user domain is identified with the name "Sh3Usr" and, clearly, it is not clickable because it is unknown by the server); the score assigned to the peptide-domain pair by the neural network predictor; the S and P values represent, respectively, the sensitivity and the precision corresponding to the score. A graphical indicator of these values is given for each derived score, as described below.
The full list of results (including those domain-peptide pairs that the server does not visualize in the output page) can be downloaded in a text format by clicking on the proper button at the top of the page. A further button allow users to submit a new query.
Score, Sensitivity and Precision
The output score assigned to each peptide-domain pair represents the result of a transformation of the neural network's output. The neural network receives in input the peptide-domain pair properly encoded (Ferraro et al. 2006), processes it by its hidden layer and produces an output that is a linear combination of the hidden sigmoid functions. Thus, the neural network output range is quite different from the required 0-1 range of a score.
In order to produce a valuable score, the neural network output undergoes a transformation by the following phases:
- Mean value and standard deviation of the output was evaluated for the training set.
- The output is therefore transformed in a z-value by subtracting its mean value and by dividing the result with its standard deviation. After the standardization the output distribution assumes a gaussian-like shape.
- The standardized output was then normalized by applying the function (Abramowitz and Stegun, 1972):
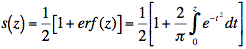
Thresholds, and corresponding Sensitivity and Precision values are reported on Table H2.
The graphical indicator
It represents the synthesis of the last two columns (sensitivity and precision): eight double-colour bars represent the eight working thresholds defined to establish the significance of the score (see Table H2). The blue and grey portions in each bar refer respectively to the Precision and Sensitivity levels, related to the threshold that the score could exceed. A white arrow indicates which bar corresponds to the peptide-domain score.
top
The SH3-Hunter engine
The background engine of SH3-Hunter is represented by a neural network predictor that receives the peptide-domain pair as input and produces a measure of the interaction propensity of the pair as output. The model was previously developed (Ferraro et al. 2006) as a structure-based model: It integrates sequence and structure information of the peptide-domain pair in two phases. The first phase consists of the appropriate selection, from peptide and domain sequences, of the amino acids directly exposed on the interaction surface. The strong conservation of SH3 domain fold assures that the exposed residues can almost always be found in 27 non-contiguous sequence positions. Such contact positions can be identified directly form complexes with known structure in the PDB or by homology modelling (Ferraro et al. 2006, Brannetti et al. 2000). For what concerns the peptides, a core of 10 contiguous positions involving the SH3 binding motifs was identified. In the second phase, Pairs of Interacting Residues (PAIRs, see Ferraro et al. 2006) are defined, consisting of pairs of amino acids, the former belonging to the 10-residues peptide, the latter belonging to one of the 27 domain contact positions. From the 270 possible residue-residue pairs, only those involving positions in physical contact are retained, thus resulting in a set of 57 PAIRs for class I binding and 53 PAIRs for class II binding.
Each peptide-domain pair can then be represented by a collections of PAIRs. The definition of the object PAIR implies a different point of view of the interaction between the domain and the peptide because it enhances the role of each domain surface residue, and in particular its coupling with each ligand residues, in the identification of the interaction specificity. Some domain surface residues could be either irrelevant when paired with some ligand residues or crucial when paired with other kinds of ligand residues. Other domain residues could also be definitively necessary for the formation of the complex, independently of the ligand residues (this is the trivial case of the conserved tryptophan in the SH3 domain sequences).
The amino acids pairs have to be properly encoded to be processed by the neural network model. Avoiding the use of high dimensional input space, the PAIRs are encoded following a knowledge-based approach that takes into account the significance of each PAIR in the interaction.
In order to identify the meaning of each PAIR, we have to evaluate their occurrences in the observed experiments of domain-peptide interactions (Landgraf et al. 2004; Tong et al. 2002).
Considering the experimental dataset used to train the SH3-Hunter neural network, the relevance of a PAIR in the formation of a complex can be assessed by examining its frequency within the binding and the non-binding peptide subsets.
Given the PAIR (p, d)k in the position k, where p and d are elements of the set {A, C, D, E, F, G, H, I, K, L, M, N, P, Q, R, S, T, V, W, Y, X}, which represents the set of all amino acids plus the insertions (X), the relative frequencies within binding and non binding subsets were defined as:
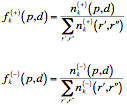
A numerical code Ck(p, d) can be proposed for a PAIR (p, d)k, according to its binding significance:
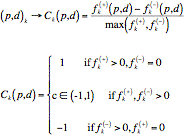
The SH3-Hunter neural network obtained outstanding performances on benchmark tests with respect to other standard model and to neural networks involving different encodings.
top
Table H1
| Domain identifier | SwissProt AC | Description |
| Nck1-SH3-1 | P16333 | Cytoplasmic protein NCK1 (2-61) |
| Nck1-SH3-2 | P16333 | Cytoplasmic protein NCK1 (115-165) |
| Nck1-SH3-3 | P16333 | Cytoplasmic protein NCK1 (190-252) |
| Nck2-SH3-1 | O43639 | Cytoplasmic protein NCK2 (2-61) |
| Nck2-SH3-2 | O43639 | Cytoplasmic protein NCK2 (111-170) |
| Nck2-SH3-3 | O43639 | Cytoplasmic protein NCK2 (195-257) |
| Obsc | Q5VST9 | Obscurin-myosin light chain kinase (5601-5668) |
| Last | - | putative protein [Arabidopsis thaliana] |
| Vav1-SH3c | P15498 | Proto-oncogene vav (617-660) |
| Vav1-SH3n | P15498 | Proto-oncogene vav (782-842) |
| Vav2-SH3c | P52735 | Protein vav-2 (586-652) |
| Vav2-SH3n | P52735 | Protein vav-2 (816-877) |
| Abo | P00519 | Proto-oncogene tyrosine-protein kinase ABL1 (61-121) |
| Fmk | P12931 | Proto-oncogene tyrosine-protein kinase Src (84-145) |
| Ad5 | P08631 | Tyrosine-protein kinase HCK (78-138) |
| Ruk-SH3-1 | Q8R550 | Regulator of ubiquitous kinase (1-58) |
| Ruk-SH3-2 | Q8R550 | Regulator of ubiquitous kinase (98-157) |
| Ruk-SH3-3 | Q8R550 | Regulator of ubiquitous kinase (311-372) |
| Tec4 | P24604 | Tyrosine-protein kinase Tec (178-238) |
| Erg13 | Q9H6S3 | Epidermal growth factor receptor kinase substrate 8-like protein 2 (492-551) |
| Erg1h | Q8TE68 | Epidermal growth factor receptor kinase substrate 8-like protein 1 (478-537) |
| Eps8h | Q12929 | Epidermal growth factor receptor kinase substrate 8 (531-590) (human) |
| Eps8m | Q08509 | Epidermal growth factor receptor kinase substrate 8 (532-591) (mouse) |
| Eps8hm | Epidermal growth factor receptor kinase substrate 8 mutated (human) | |
| Neb | P20929 | Nebulin (6610-6669) |
| Cka | P46108 | Proto-oncogene C-crk (132-192) (human, mouse, rattus) |
| Crk | Q04929 | Proto-oncogene C-crk (133-193) (chicken) |
| Efn | P06241 | Proto-oncogene tyrosine-protein kinase Fyn (82-143) |
| Spc | Q13813 | Spectrin alpha chain, brain (967-1026) (SPTA2_Human) |
| Fyn | P06241 | Proto-oncogene tyrosine-protein kinase Fyn (82-143) |
| Yes | P07947 | Proto-oncogene tyrosine-protein kinase Yes (91-152) |
| P85A | P27986 | PI3-kinase p85-subunit alpha (3-79) |
| Itk | Q08881 | Tyrosine-protein kinase ITK/TSK (171-231) |
| Lyn | P07948 | Tyrosine-protein kinase Lyn (63-123) |
| P53bp | Q13625 | p53-binding protein 2 (1057-1119) |
| Sem5 | P29355 | Sex muscle abnormal protein 5 (154-213) |
| Grb-n | P62993 | Growth factor receptor-bound protein 2 (1-58) |
| Grb-c | P62993 | Growth factor receptor-bound protein 2 (156-215) |
| Plc | P19174 | Phospholipase C-gamma-1 (791-851) |
| Cor | Q60598 | Src substrate cortactin (488-546) |
| Amp-c | P50478 | Amphiphysin (609-682) (chicken) |
| Amp-h | P49418 | Amphiphysin (622-695) (human) |
| End3 | Q99963 | SH3-containing GRB2-like protein 3 (285-344) |
| End2 | Q99962 | SH3-containing GRB2-like protein 2 (290-349) |
| End1 | Q99961 | SH3-containing GRB2-like protein 1 (306-365) |
| Myo3 | P36006 | Myosin-3 (1121-1183) |
| Myo5 | Q04439 | Myosin-5 (1085-1147) |
| Bem1-1 | P29366 | Bud emergence protein 1 (72-132) |
| Bem1-2 | P29366 | Bud emergence protein 1 (155-217) |
| Nbp2 | Q12163 | NAP1-binding protein 2 (110-171) |
| Abp1 | P15891 | Actin-binding protein (532-592) |
| Ydl117 | Q07533 | Cytokinesis protein 3 (9-70) |
| Sla1-1 | P32790 | Cytoskeleton assembly control protein SLA1 (8-69) |
| Sla1-2 | P32790 | Cytoskeleton assembly control protein SLA1 (70-132) |
| Sla1-3 | P32790 | Cytoskeleton assembly control protein SLA1 (353-415) |
| Ygr136 | P53281 | LAS17-interacting protein 1 (53-112) |
| Ypr154 | Q06449 | LAS17-binding protein 2 (54-113) |
| Yhl002 | P38753 | Uncharacterized protein YHL002W (217-276) |
| Yhr114-1 | P38822 | Protein BZZ1 (493-555) |
| Yhr114-2 | P38822 | Protein BZZ1 (577-633) |
| Yar014 | P27637 | Bud site selection protein 14 (259-320) |
| Fus1 | P11710 | Nuclear fusion protein FUS1 (436-512) |
| Yll017 | P14771 | Guanine nucleotide exchange factor SDC25 (26-97) |
| Cdc25 | P04821 | Cell division control protein 25 (58-128) |
| Sho1 | P40073 | Protein SSU81 (300-361) |
| Yhr016 | P32793 | Protein YSC84 (409-468) |
| Rvs167 | P39743 | Reduced viability upon starvation protein 167 (421-482) |
| Ymr032 | Q05080 | Cytokinesis 2 protein (599-667) |
| Yjl020 | P47068 | Protein BBC1 (5-69) |
| Yfr024 | P43603 | LAS17-binding protein 3 (392-451) |
| Boi1 | P38041 | Protein BOB1 (13-77) |
| Boi2 | P39969 | Protein BEB1 (43-107) |
| Pex13 | P80667 | Peroxin-13 (306-372) |
Table H2
| Class I | Class II | ||||
| Threshold | Sensitivity | Precision | Threshold | Sensitivity | Precision |
| 0.95 | 98% | 50% | 0.9 | 98% | 50% |
| 0.97 | 95% | 60% | 0.94 | 93% | 60% |
| 0.98 | 92% | 70% | 0.96 | 87% | 70% |
| 0.99 | 83% | 80% | 0.98 | 81% | 80% |
| 0.994 | 73% | 90% | 0.995 | 64% | 90% |
| 0.998 | 64% | 95% | 0.9997 | 54% | 95% |
| 0.999 | 60% | 97% | 0.9999 | 50% | 97% |
| 1 | 17% | 100% | 1 | 23% | 100% |
- Ferraro, E., Peluso, D., Via, A., Ausiello, G., Helmer-Citterich, M., (2007) SH3-Hunter: discovery of SH3 domain interaction sites in proteins. Nucleic Acids Res. 35 (Web Server issue): W451-4. paper
- Abramowitz, M. and Stegun, I. A. (Eds.). "Error Function and Fresnel Integrals." Ch. 7 in Handbook of Mathematical Functions with Formulas, Graphs, and Mathematical Tables, 9th printing. New York: Dover, pp. 297-309, 1972.
- Brannetti, B., Via, A., Cestra, G., Cesareni, G., and Helmer-citterich, M., (2000) SH3-SPOT: An algorithm to predict preferred ligands to different members of the SH3 gene family. J. Mol. Biol. 298, 313-328.
- Ferraro, E., Via, A., Ausiello, G., Helmer-Citterich, M., (2006) A novel structure-based encoding for machine-learning applied to the inference of SH3 domain specificity. Bioinformatics 22(19), 2333-2339.
- Landgraf, C., et al, (2004) Protein interaction networks by proteome peptide scanning. PLOS Biol. 2, 94-103.
- Tong, A., H., et al, (2002) A combined experimental and computational strategy to define protein interaction networks for peptide recognition modules. Science 295, 321-324.